Building more complex Seurat objects #
In most cases, we will want to go beyond reading one sample into Seurat and performing biological analyses. We often want to create a Seurat object that has multiple samples (e.g. biological replicates or patients). These samples will also have unique pieces of metadata, such as HPV- PBMC or HPV+ TIL from our head and neck cancer dataset.
Prerequisites #
This analysis assumes that you have download the samples described in the GEO download tutorial under utilities.
Load packages #
First, we will load the necessary packages.
library(Seurat)
## Attaching SeuratObject
library(dplyr)
##
## Attaching package: 'dplyr'
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
library(patchwork)
Read the individual files into a list #
The data I downloaded using the tutorial are located in /home/rstudio/docker_rstudio/data/geo_download. We will create a simple for loop to read all the files into individual variables stored in a list.
full_path <- "/home/rstudio/docker_rstudio/data/geo_download/"
raw_files <- list.files(paste(full_path))
# Not the metadata... we will use this in a second
raw_files <- raw_files[2:5]
raw_files
## [1] "HD_PBMC_1" "HD_PBMC_2" "HD_PBMC_3" "HD_PBMC_4"
# Create list for Seurat object
raw_list <- vector("list",length=length(raw_files))
raw_list # empty list of length 4
## [[1]]
## NULL
##
## [[2]]
## NULL
##
## [[3]]
## NULL
##
## [[4]]
## NULL
# Loop to read filtered feature barcode matrices into R data into R
for (i in 1:length(raw_list)) {
raw_list[[i]] <- Read10X(paste(full_path,raw_files[i],sep=""))
}
dim(raw_list[[1]]) # 33694 genes by 2445 cells
## [1] 33694 2445
raw_list[[1]][1:50,1:10] # sparse matrix with counts
## 50 x 10 sparse Matrix of class "dgCMatrix"
## [[ suppressing 10 column names 'AAACCTGCACAGACTT-1', 'AAACCTGCATCGGTTA-1', 'AAACCTGTCAAGCCTA-1' ... ]]
##
## RP11-34P13.3 . . . . . . . . . .
## FAM138A . . . . . . . . . .
## OR4F5 . . . . . . . . . .
## RP11-34P13.7 . . . . . . . . . .
## RP11-34P13.8 . . . . . . . . . .
## RP11-34P13.14 . . . . . . . . . .
## RP11-34P13.9 . . . . . . . . . .
## FO538757.3 . . . . . . . . . .
## FO538757.2 . . . . . . 1 . . .
## AP006222.2 1 . . . . . . . . .
## RP5-857K21.15 . . . . . . . . . .
## RP4-669L17.2 . . . . . . . . . .
## RP4-669L17.10 . . . . . . . . . .
## OR4F29 . . . . . . . . . .
## RP5-857K21.4 . . . . . . . . . .
## RP5-857K21.2 . . . . . . . . . .
## OR4F16 . . . . . . . . . .
## RP11-206L10.4 . . . . . . . . . .
## RP11-206L10.9 . . . . . . . . . .
## FAM87B . . . . . . . . . .
## LINC00115 . . . . . . . . . .
## FAM41C . . . . . . . . . .
## RP11-54O7.16 . . . . . . . . . .
## RP11-54O7.1 . . . . . . . . . .
## RP11-54O7.2 . . . . . . . . . .
## RP11-54O7.3 . . . . . . . . . .
## SAMD11 . . . . . . . . . .
## NOC2L . . . 2 . . . . . .
## KLHL17 . . . . . . . . . .
## PLEKHN1 . . . . . . . . . .
## PERM1 . . . . . . . . . .
## RP11-54O7.17 . . . . . . . . . .
## HES4 . . . . . . . . . .
## ISG15 . 4 1 . . . 1 1 . .
## RP11-54O7.11 . . . . . . . . . .
## AGRN . . . . . . . . . .
## RP11-54O7.18 . . . . . . . . . .
## RNF223 . . . . . . . . . .
## C1orf159 . . . . . . . . . .
## LINC01342 . . . . . . . . . .
## RP11-465B22.8 . . . . . . . . . .
## TTLL10-AS1 . . . . . . . . . .
## TTLL10 . . . . . . . . . .
## TNFRSF18 . . . . . . . . . .
## TNFRSF4 . . . 1 1 . . 1 . .
## SDF4 . . . . . . . 1 . .
## B3GALT6 . . . . . . . . . .
## FAM132A . . . . . . . . . .
## RP5-902P8.12 . . . . . . . . . .
## UBE2J2 . . . . . . . . . .
# Name each item in the list by its title
names(raw_list) <- raw_files
names(raw_list)
## [1] "HD_PBMC_1" "HD_PBMC_2" "HD_PBMC_3" "HD_PBMC_4"
Read in metadata #
Before we construct the combined object, we will need the metadata.
We can read that in from the tsv file using readr::read_tsv (readr is a package, and the “::” lets us call a function from that package without loading the package)
# Read in the metadata
metadata_file <- readr::read_tsv("/home/rstudio/docker_rstudio/data/geo_download/GSE139324_metadata.tsv")
## Rows: 63 Columns: 45
## ── Column specification ────────────────────────────────────────────────────────
## Delimiter: "\t"
## chr (41): sample_accession, title, geo_accession, status, submission_date, l...
## dbl (4): channel_count, taxid_ch1, contact_zip/postal_code, data_row_count
##
## ℹ Use `spec()` to retrieve the full column specification for this data.
## ℹ Specify the column types or set `show_col_types = FALSE` to quiet this message.
# Re-name a few columns for easy use
colnames(metadata_file)[c(43,45)] <- c("disease_state","tissue")
# Filter to only our samples of interest
metadata_file_filtered <- metadata_file %>%
filter(title %in% raw_files)
# Make sure the order of names matches the raw data list
identical(metadata_file_filtered$title,names(raw_list))
## [1] TRUE
# Create an empty list of Seurat objects
ser_list <- vector("list",length=length(raw_list))
# Loop to Create individual Seurat objects and include metadata
for (i in 1:length(ser_list)) {
# Create Seurat Object
ser_list[[i]] <- CreateSeuratObject(raw_list[[i]])
# Create metadata for each object
meta_add <- data.frame(matrix(data=NA,nrow=ncol(ser_list[[i]]),ncol=3))
colnames(meta_add) <- c("title","disease_state","tissue")
rownames(meta_add) <- colnames(ser_list[[i]])
meta_add$title <- metadata_file_filtered[i,"title"] %>% pull()
meta_add$disease_state <- metadata_file_filtered[i,"disease_state"] %>% pull()
meta_add$tissue <- metadata_file_filtered[i,"tissue"] %>% pull()
# Add Metadata
ser_list[[i]] <- AddMetaData(ser_list[[i]],metadata=meta_add)
}
## Warning: Feature names cannot have underscores ('_'), replacing with dashes
## ('-')
## Warning: Feature names cannot have underscores ('_'), replacing with dashes
## ('-')
## Warning: Feature names cannot have underscores ('_'), replacing with dashes
## ('-')
## Warning: Feature names cannot have underscores ('_'), replacing with dashes
## ('-')
# Check out our list of Seurat objects
ser_list
## [[1]]
## An object of class Seurat
## 33694 features across 2445 samples within 1 assay
## Active assay: RNA (33694 features, 0 variable features)
##
## [[2]]
## An object of class Seurat
## 33694 features across 2436 samples within 1 assay
## Active assay: RNA (33694 features, 0 variable features)
##
## [[3]]
## An object of class Seurat
## 33694 features across 1767 samples within 1 assay
## Active assay: RNA (33694 features, 0 variable features)
##
## [[4]]
## An object of class Seurat
## 33694 features across 2315 samples within 1 assay
## Active assay: RNA (33694 features, 0 variable features)
Merge list of Seurat objects into a single seurat object #
ser_merged <- merge(ser_list[[1]],ser_list[2:4])
## Warning in CheckDuplicateCellNames(object.list = objects): Some cell names are
## duplicated across objects provided. Renaming to enforce unique cell names.
ser_merged@meta.data$title %>%
table()
## .
## HD_PBMC_1 HD_PBMC_2 HD_PBMC_3 HD_PBMC_4
## 2445 2436 1767 2315
ser_merged@meta.data %>%
select(title,disease_state) %>%
table()
## disease_state
## title Healthy donor
## HD_PBMC_1 2445
## HD_PBMC_2 2436
## HD_PBMC_3 1767
## HD_PBMC_4 2315
ser_merged@meta.data %>%
select(title,tissue) %>%
table()
## tissue
## title peripheral blood
## HD_PBMC_1 2445
## HD_PBMC_2 2436
## HD_PBMC_3 1767
## HD_PBMC_4 2315
Analysis of all 4 samples #
As per the shortcut in the PBMC vignette, here’s a quick analysis of the 4 healthy donor samples we merged into one Seurat object.
ser_merged <- ser_merged %>%
NormalizeData() %>%
FindVariableFeatures() %>%
ScaleData() %>%
RunPCA()
## Centering and scaling data matrix
## PC_ 1
## Positive: RPS29, IL32, TRAC, LTB, RPS18, TRBC2, IL7R, RPS12, TRBC1, RPS2
## EEF1A1, CCR7, CD247, CD69, PEBP1, TRAT1, GZMM, MYC, AQP3, TSHZ2
## CTSW, FHIT, NGFRAP1, CDC25B, RPS26, HIST1H4C, FKBP11, NELL2, CD8B, CD40LG
## Negative: CST3, FCN1, LYZ, CSTA, LST1, S100A9, CTSS, TYROBP, S100A8, MNDA
## FGL2, FTL, SAT1, TYMP, FTH1, VCAN, PSAP, FOS, LGALS1, FCER1G
## S100A12, SERPINA1, CFD, CYBB, NEAT1, SPI1, MS4A6A, AIF1, HLA-DRA, GPX1
## PC_ 2
## Positive: LTB, CCR7, RPS12, RPS18, EEF1A1, RPS2, RPLP1, CD79A, MYC, MS4A1
## IGHM, IL7R, NCF1, IGHD, LINC00926, BANK1, BIRC3, VPREB3, RPS29, FHIT
## TSHZ2, TCL1A, VIM, CD22, IGKC, TNFRSF13C, NGFRAP1, TRAC, FCRLA, FCER2
## Negative: NKG7, CST7, GZMA, PRF1, GNLY, KLRD1, GZMB, FGFBP2, HOPX, CTSW
## KLRF1, CCL5, SPON2, GZMH, TRDC, FCGR3A, CLIC3, CCL4, MATK, KLRB1
## ADGRG1, MYO1F, PFN1, S1PR5, C12orf75, CMC1, IL2RB, TTC38, PRSS23, C1orf21
## PC_ 3
## Positive: TRAC, IL7R, IL32, VIM, S100A6, S100A12, VCAN, S100A8, ANXA1, S100A9
## CXCL8, TRAT1, S100A4, TRBC1, MCL1, CD14, CH17-373J23.1, RGCC, LYZ, IL1B
## AIF1, NEAT1, FHIT, RBP7, NFKBIZ, RP11-160E2.6, TSHZ2, CSTA, DUSP6, TSPO
## Negative: CD79A, MS4A1, IGHM, CD79B, IGKC, LINC00926, IGHD, BANK1, TCL1A, VPREB3
## SPIB, HLA-DQA1, CD22, FAM129C, BLNK, HLA-DPA1, CD74, FCRLA, HLA-DPB1, FCER2
## TSPAN13, HLA-DOB, HLA-DQB1, TNFRSF13C, IGLC2, BLK, JCHAIN, CD19, CD24, RP11-693J15.5
## PC_ 4
## Positive: CDKN1C, HES4, CKB, CSF1R, TCF7L2, MS4A4A, SIGLEC10, CTD-2006K23.1, HMOX1, FCGR3A
## MS4A7, VMO1, BATF3, LINC01272, LRRC25, ZNF703, IFITM3, FAM110A, LYPD2, ICAM4
## CTSL, CDH23, TPPP3, RHOC, C1QA, LILRB2, PILRA, BID, OAS1, CD68
## Negative: CXCL8, VCAN, S100A12, IL1B, RP11-160E2.6, CH17-373J23.1, NFKBIA, FOSB, CCL3, FOS
## S100A8, IER3, CTD-3252C9.4, TNFAIP3, JUN, RP11-1143G9.4, EGR1, NCF1, JUND, LUCAT1
## S100A9, ITGAM, CD14, LINC00936, MT-CO1, MCL1, MS4A6A, KLF10, CYP1B1, CLEC4E
## PC_ 5
## Positive: CD79B, MS4A1, CD79A, LINC00926, IGHD, FCGR3A, CDKN1C, VPREB3, CD22, FCER2
## BANK1, MS4A7, SIGLEC10, IGHM, HES4, FCRL1, TNFRSF13C, CKB, MTSS1, TCF7L2
## LINC01272, HLA-DOB, FCRLA, RP11-693J15.5, CD19, CD24, RALGPS2, CD72, SWAP70, HMOX1
## Negative: SERPINF1, PLD4, GAS6, LRRC26, PPP1R14B, CLEC4C, LILRA4, ITM2C, TPM2, FCER1A
## DERL3, SCT, PTCRA, SMPD3, C1orf186, JCHAIN, LINC00996, SCN9A, IL3RA, LILRB4
## PTPRS, CCDC50, RP11-117D22.2, MZB1, UGCG, RP11-73G16.2, DNASE1L3, LAMP5, MAP1A, TNFRSF21
ElbowPlot(ser_merged)
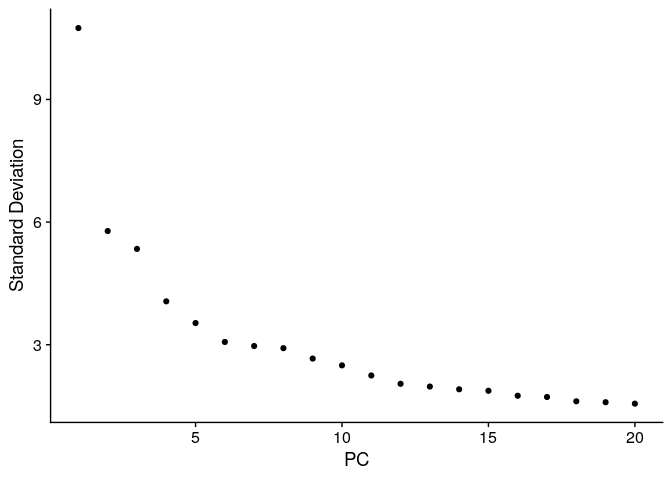
ser_merged <- ser_merged %>%
FindNeighbors(.,dims=1:15) %>%
FindClusters(.,res=c(0.3,0.5,0.7)) %>%
RunUMAP(.,dims=1:10)
## Computing nearest neighbor graph
## Computing SNN
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 8963
## Number of edges: 319663
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.9303
## Number of communities: 13
## Elapsed time: 0 seconds
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 8963
## Number of edges: 319663
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.9083
## Number of communities: 16
## Elapsed time: 0 seconds
## Modularity Optimizer version 1.3.0 by Ludo Waltman and Nees Jan van Eck
##
## Number of nodes: 8963
## Number of edges: 319663
##
## Running Louvain algorithm...
## Maximum modularity in 10 random starts: 0.8883
## Number of communities: 17
## Elapsed time: 0 seconds
## Warning: The default method for RunUMAP has changed from calling Python UMAP via reticulate to the R-native UWOT using the cosine metric
## To use Python UMAP via reticulate, set umap.method to 'umap-learn' and metric to 'correlation'
## This message will be shown once per session
## 19:09:30 UMAP embedding parameters a = 0.9922 b = 1.112
## 19:09:30 Read 8963 rows and found 10 numeric columns
## 19:09:30 Using Annoy for neighbor search, n_neighbors = 30
## 19:09:30 Building Annoy index with metric = cosine, n_trees = 50
## 0% 10 20 30 40 50 60 70 80 90 100%
## [----|----|----|----|----|----|----|----|----|----|
## **************************************************|
## 19:09:30 Writing NN index file to temp file /tmp/RtmpZYOKxd/file44aa2cf3936a
## 19:09:30 Searching Annoy index using 1 thread, search_k = 3000
## 19:09:32 Annoy recall = 100%
## 19:09:32 Commencing smooth kNN distance calibration using 1 thread with target n_neighbors = 30
## 19:09:33 Initializing from normalized Laplacian + noise (using irlba)
## 19:09:33 Commencing optimization for 500 epochs, with 368458 positive edges
## 19:09:38 Optimization finished
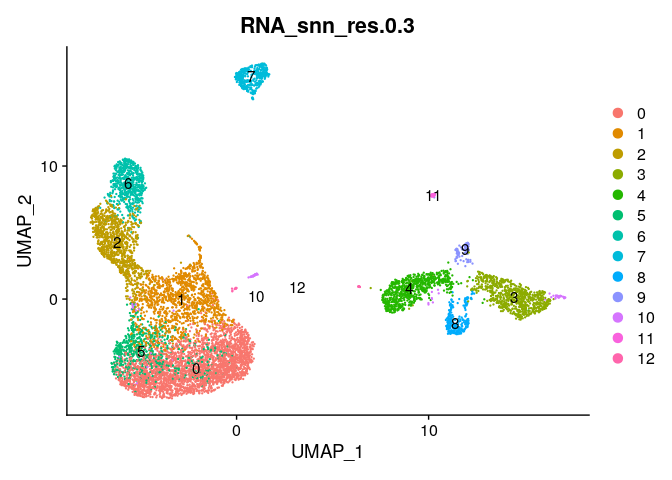
Plot a UMAP of clusters by sample #
DimPlot(ser_merged,group.by="RNA_snn_res.0.3",label=T)
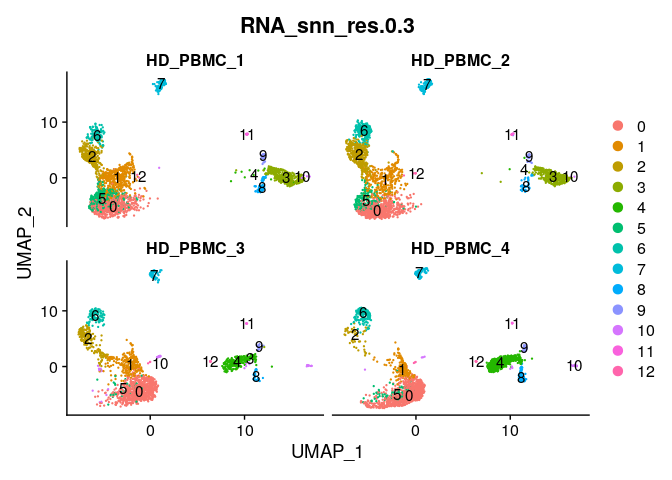
DimPlot(ser_merged,group.by="RNA_snn_res.0.3",split.by="title",label=T,
ncol=2)
Save output #
save_path <- "/home/rstudio/docker_rstudio/data/"
saveRDS(ser_merged,file=paste(save_path,"ser_merged_4_hd_pbmc_231027.rds",sep=""))